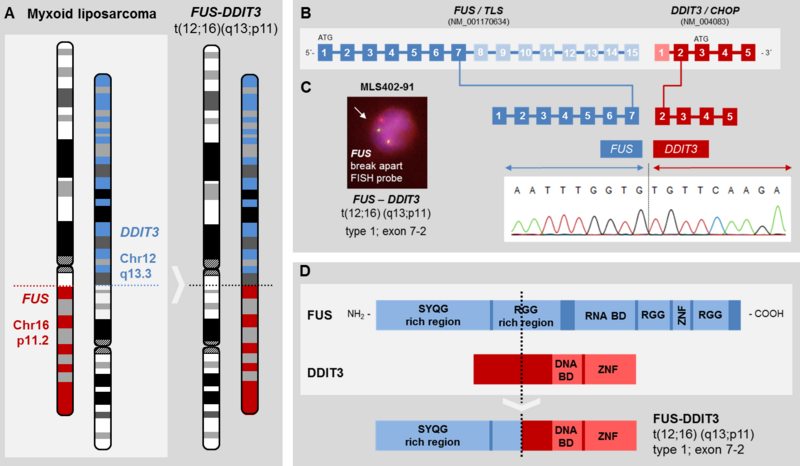
Synovial sarcomas (SySa) account for 5-10% of all soft tissue sarcomas. In the majority, SySa arise mainly in adolescents and young adults with predominance in male gender. They are molecularly characterized by a reciprocal t(X;18) translocation which juxtaposes the SS18 gene on chromosome 18 to either the SSX1, the SSX2, or rarely to the SSX4 gene on the X chromosome. The SS18-SSX chimeric proteins act as transcriptional co-activators, leading to deregulation of oncogenic pathways. Current treatment protocols for synovial sarcoma are based on radical surgery and standardized chemo- and radiotherapy; however, prognosis is still poor in advanced disease. Targeted therapeutic approaches, which have significantly improved the clinical course of patients with e.g. GIST or dermato- fibrosarcoma protuberans, are still lacking for SySa. Several receptor tyrosine kinases have been shown to be expressed in SySa, including the EGF receptor, and the IGF-IR, leading to an activation of the PI3K/AKT signaling pathway. Beyond kinase signals, we have shown an essential role of WNT/ß-catenin signals in SySa.
Our current studies analyze the functional relevance of diverse oncogenic signaling pathways in synovial sarcoma, aiming at a better understanding of tumor biology as a basis for the definition of innovative therapeutic approaches.