Leitung: OA Dr. Volker Debus
Zu den genetisch basierten elektrischen Herzerkrankungen, die mit einem erhöhten Risiko für Herzrhythmusstörrungen (ventrikuläre Tachykardien) und einen plötzlichen Herztod einhergehen gehören das/die:
· Langes QT-Syndrom, Long QT-Syndrom (LQTS)
· Brugada-Syndrom (BrS)
· Katecholaminerge, polymorphe ventrikuläre Tachykardien (CPVT)
· Arhythmogene rechtsventrikuläre Dysplasie (ARVD)
· Kurzes QT-Syndrom (SQTS)
· Progressive kardiale Leitungsverzögerung (PCCD)
In unserer Spezialsprechstunde betreuen wir Kinder und Jugendliche, bei denen eine derartige Erkrankung vermutet wird oder bereits bestätigt wurde. Neben der eingehenden und umfassenden Untersuchung und diagnostischen Abklärung stehen dabei eine adäquate Behandlung und die Beratung der betroffenen Familien im Zentrum unserer Arbeit.
Dabei arbeiten wir am Universitätsklinikum Münster eng mit dem hiesigen Institut für Genetik von Herzerkrankungen (Direktor Univ.-Prof. E. Schulze-Bahr) zusammen. Aufgrund der herausragenden Expertise in diesem Fachgebiet erhält unsere Spezialambulanz mittlerweile weit überregionale Zuweisungen aus dem gesamten Bundesgebiet.
Long QT-Syndrom (LQTS)
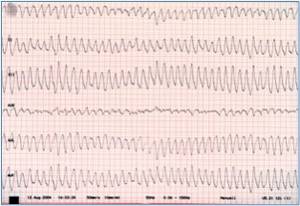
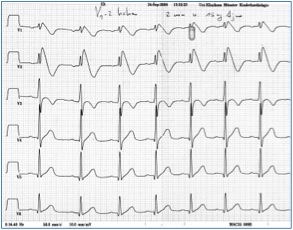
Das Syndrom der verlängerten QT-Zeit im EKG (LQTS) ist eine Gruppe von ähnlichen Ionenkanal-Erkrankungen des Herzens, die mit einer erhöhten Neigung zu Bewußtlosigkeiten (Synkopen) und Herzrhythmusstörungen (insb. sog. Torsade de pointes (TdP) oder Spitzenumkehrtachykardien, Abb.1) einhergehen. Meistens sistiert dieses Herzrasen nach kurzer Zeit spontan, es kann aber auch Minuten anhalten und im schlimmsten Fall zum plötzlichen Herztod führen.
Die Häufigkeit dieser oft angeborenen und vererbten Erkrankung wird auf 1:1200 geschätzt. Eine gründliche Erhebung der Eigen- und Familienvorgeschichte (Eigen- und Familien-Anamnese) ist daher neben dem Ruhe-EKG die Grundlage der Diagnostik.
Die häufigste Form des angeborenen LQTS (Romano-Ward-Syndrom) wird autosomal dominant vererbt. Dies bedeutet, dass jedes Kind eines betroffen/erkrankten Elternteils eine Wahrscheinlichkeit von 50% besitzt, ebenfalls von dieser genetischen Veränderung betroffen zu sein.
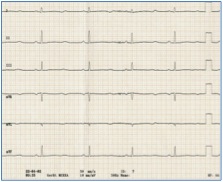
Eine seltenere Form des angeborenen LQTS (Jervell-Lange-Nielsen-Syndrom, Abb.2) wird autosomal rezessiv vererbt. Dies bedeutet, dass jedes Kind beider betroffen/erkrankten Elternteile eine Wahrscheinlichkeit von 25% besitzt, ebenfalls von dieser genetischen Veränderung betroffen zu sein.
In 70-80% der LQTS-Fälle lässt sich molekulargenetisch eine Veränderung (Mutation) in den Genen nachweisen, die für den Aufbau von Ionenkanälen im Herzen verantwortlich sind. Bestimmte Ionenkanäle (IKs, IKr, INa) werden dann fehlerhaft aufgebaut. Mittlerweile sind mindestens 13 LQTS-Unterformen bekannt, wobei LQTS 1-3 für ca. 90% aller genetisch bestätigten Patienten verantwortlich zeichnen.
Diese Molekulargenetik wird am UKM im hiesigen Institut für Genetik von Herzerkrankungen unter Leitung von Univ.-Prof. E. Schulze-Bahr durchgeführt. Mit entsprechender Überweisung (Laborschein 10, Molekulargenetik) und nach ausführlicher Beratung beider sorgeberechtigten Eltern nach dem Gen-Diagnostikgesetz (GenDG) nehmen wir EDTA-Blut zur entsprechenden Diagnostik ab.
Nach der Diagnosestellung eines Long QT-Syndroms sind folgende Maßnahmen zu empfehlen:
1. Vermeidung von Triggeraktivitäten wie z.B.:
- Wettkampfsport, sportliche Höchstleistungen
- Sprung ins kalte Wasser, unbeaufsichtigtes Schwimmen, Tauchen
- Schrecksituationen, Wecker/Telefon/Handy im Schlafraum
- Meidung weiterer repolarisationsverlängernder Medikamente
- Elektrolytmangel (Kalium, Magnesium) durch anhaltendes
Erbrechen, Durchfall, Anorexie, Rachitis
2. Medikamente
- ß-Blocker sind bei allen Formen wirksam
- Mexiletin bei LQT3
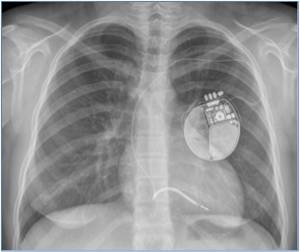
3. Defibrillator, ICD
- Indikation bei überlebtem plötzlichem Herztod als
Sekundärprophylaxe (Klasse I)
- Indikation bei Ereignissen (Kammertachykardien / Synkopen)
unter adäquater Medikation als Primärprophylaxe (Klasse IIA) (s.Abb.3)
Brugada-Syndrom (BrS)
Das Brugada-Syndrom ist eine primär elektrische Herzerkrankung (Na-Ionenkanalopathie) bei sonst strukturell unauffälligem Herzen und wurde erstmals 1992 von den Brüdern Pedro und Josep Brugada beschrieben.
Die Häufigkeit dieser oft angeborenen (autosomal dominant mit unterschiedlicher Penetranz) und vererbten Erkrankung wird auf 1-5:10.000 in westlichen und auf 1:2.500 in fernöstlichen Ländern geschätzt.
Eine gründliche Erhebung der Eigen- und Familienvorgeschichte (Eigen- und Familien-Anamnese) ist daher neben dem Ruhe-EKG und ggf. Provokations-EKG die Grundlage der Diagnostik.
In ca. 20% der Fälle lässt sich molekulargenetisch eine Veränderung (Mutation) im Gen SCN5A nachweisen, das für den Aufbau des Na-Ionenkanals im Herzen verantwortlich ist.
Diese Molekulargenetik wird am UKM im hiesigen Institut für Genetik von Herzerkrankungen unter Leitung von Univ.-Prof. E. Schulze-Bahr durchgeführt. Mit entsprechender Überweisung (Laborschein 10, Molekulargenetik) und nach ausführlicher Beratung beider sorgeberechtigten Eltern nach dem Gen-Diagnostikgesetz (GenDG) nehmen wir EDTA-Blut zur entsprechenden Diagnostik ab.
Zu den genetisch basierten elektrischen Herzerkrankungen, die mit einem erhöhten Risiko für Herzrhythmusstörrungen (ventrikuläre Tachykardien) und einen plötzlichen Herztod einhergehen gehören das/die:
· Langes QT-Syndrom, Long QT-Syndrom (LQTS)
· Brugada-Syndrom (BrS)
· Katecholaminerge, polymorphe ventrikuläre Tachykardien (CPVT)
· Arhythmogene rechtsventrikuläre Dysplasie (ARVD)
· Kurzes QT-Syndrom (SQTS)
· Progressive kardiale Leitungsverzögerung (PCCD)
In unserer Spezialsprechstunde betreuen wir Kinder und Jugendliche, bei denen eine derartige Erkrankung vermutet wird oder bereits bestätigt wurde. Neben der eingehenden und umfassenden Untersuchung und diagnostischen Abklärung stehen dabei eine adäquate Behandlung und die Beratung der betroffenen Familien im Zentrum unserer Arbeit.
Dabei arbeiten wir am Universitätsklinikum Münster eng mit dem hiesigen Institut für Genetik von Herzerkrankungen (Direktor Univ.-Prof. E. Schulze-Bahr) zusammen. Aufgrund der herausragenden Expertise in diesem Fachgebiet erhält unsere Spezialambulanz mittlerweile weit überregionale Zuweisungen aus dem gesamten Bundesgebiet.
Long QT-Syndrom (LQTS)
Das Syndrom der verlängerten QT-Zeit im EKG (LQTS) ist eine Gruppe von ähnlichen Ionenkanal-Erkrankungen des Herzens, die mit einer erhöhten Neigung zu Bewußtlosigkeiten (Synkopen) und Herzrhythmusstörungen (insb. sog. Torsade de pointes (TdP) oder Spitzenumkehrtachykardien, Abb.1) einhergehen. Meistens sistiert dieses Herzrasen nach kurzer Zeit spontan, es kann aber auch Minuten anhalten und im schlimmsten Fall zum plötzlichen Herztod führen.
Die Häufigkeit dieser oft angeborenen und vererbten Erkrankung wird auf 1:1200 geschätzt. Eine gründliche Erhebung der Eigen- und Familienvorgeschichte (Eigen- und Familien-Anamnese) ist daher neben dem Ruhe-EKG die Grundlage der Diagnostik.
Die häufigste Form des angeborenen LQTS (Romano-Ward-Syndrom) wird autosomal dominant vererbt. Dies bedeutet, dass jedes Kind eines betroffen/erkrankten Elternteils eine Wahrscheinlichkeit von 50% besitzt, ebenfalls von dieser genetischen Veränderung betroffen zu sein.
Eine seltenere Form des angeborenen LQTS (Jervell-Lange-Nielsen-Syndrom, Abb.2) wird autosomal rezessiv vererbt. Dies bedeutet, dass jedes Kind beider betroffen/erkrankten Elternteile eine Wahrscheinlichkeit von 25% besitzt, ebenfalls von dieser genetischen Veränderung betroffen zu sein.
In 70-80% der LQTS-Fälle lässt sich molekulargenetisch eine Veränderung (Mutation) in den Genen nachweisen, die für den Aufbau von Ionenkanälen im Herzen verantwortlich sind. Bestimmte Ionenkanäle (IKs, IKr, INa) werden dann fehlerhaft aufgebaut. Mittlerweile sind mindestens 13 LQTS-Unterformen bekannt, wobei LQTS 1-3 für ca. 90% aller genetisch bestätigten Patienten verantwortlich zeichnen.
Diese Molekulargenetik wird am UKM im hiesigen Institut für Genetik von Herzerkrankungen unter Leitung von Univ.-Prof. E. Schulze-Bahr durchgeführt. Mit entsprechender Überweisung (Laborschein 10, Molekulargenetik) und nach ausführlicher Beratung beider sorgeberechtigten Eltern nach dem Gen-Diagnostikgesetz (GenDG) nehmen wir EDTA-Blut zur entsprechenden Diagnostik ab.
Nach der Diagnosestellung eines Long QT-Syndroms sind folgende Maßnahmen zu empfehlen:
1. Vermeidung von Triggeraktivitäten wie z.B.:
- Wettkampfsport, sportliche Höchstleistungen
- Sprung ins kalte Wasser, unbeaufsichtigtes Schwimmen, Tauchen
- Schrecksituationen, Wecker/Telefon/Handy im Schlafraum
- Meidung weiterer repolarisationsverlängernder Medikamente
- Elektrolytmangel (Kalium, Magnesium) durch anhaltendes
Erbrechen, Durchfall, Anorexie, Rachitis
2. Medikamente
- ß-Blocker sind bei allen Formen wirksam
- Mexiletin bei LQT3
3. Defibrillator, ICD
- Indikation bei überlebtem plötzlichem Herztod als
Sekundärprophylaxe (Klasse I)
- Indikation bei Ereignissen (Kammertachykardien / Synkopen)
unter adäquater Medikation als Primärprophylaxe (Klasse IIA) (s.Abb.3)
Brugada-Syndrom (BrS)
Das Brugada-Syndrom ist eine primär elektrische Herzerkrankung (Na-Ionenkanalopathie) bei sonst strukturell unauffälligem Herzen und wurde erstmals 1992 von den Brüdern Pedro und Josep Brugada beschrieben.
Die Häufigkeit dieser oft angeborenen (autosomal dominant mit unterschiedlicher Penetranz) und vererbten Erkrankung wird auf 1-5:10.000 in westlichen und auf 1:2.500 in fernöstlichen Ländern geschätzt.
Eine gründliche Erhebung der Eigen- und Familienvorgeschichte (Eigen- und Familien-Anamnese) ist daher neben dem Ruhe-EKG und ggf. Provokations-EKG die Grundlage der Diagnostik.
In ca. 20% der Fälle lässt sich molekulargenetisch eine Veränderung (Mutation) im Gen SCN5A nachweisen, das für den Aufbau des Na-Ionenkanals im Herzen verantwortlich ist.
Diese Molekulargenetik wird am UKM im hiesigen Institut für Genetik von Herzerkrankungen unter Leitung von Univ.-Prof. E. Schulze-Bahr durchgeführt. Mit entsprechender Überweisung (Laborschein 10, Molekulargenetik) und nach ausführlicher Beratung beider sorgeberechtigten Eltern nach dem Gen-Diagnostikgesetz (GenDG) nehmen wir EDTA-Blut zur entsprechenden Diagnostik ab.