Morbus Fabry zählt zu den "lysosomalen Speicherkrankheiten". Es sind mehr als 50 verschiedene dieser seltenen "Stoffwechselkrankheiten" bekannt, von denen weltweit jeweils nur ein paar Tausend Menschen betroffen sind. Alle Speicherkrankheiten sind angeboren und werden durch einen "Mangel spezifischer Eiweißstoffe (Enzyme)" in den Lysosomen - den "Müllverbrennungsanlagen" der Zellen - verursacht. "Lysosomen" sind Organellen (Zellkörperchen), die mit Ausnahme der roten Blutkörperchen in allen menschlichen Körperzellen vorkommen. Die Enzyme in den Lysosomen verdauen Zucker, Eiweißstoffe, Nukleinsäuren und Fette. Fehlt eines dieser Enzyme oder wird es nicht in ausreichender Menge produziert, werden die Lysosomen mit Abfallstoffen überfüllt und es entwickelt sich eine Speicherkrankheit.
Bei Morbus Fabry handelt es sich bei dem fehlenden bzw. "mangelnden Enzym" um die so genannte "α-Galaktosidase A (α-GAL)". Dieses Enzym spaltet Fettstoffe (Glykosphingolipide), die wichtige Bestandteile der Hülle unserer Körperzellen sind. Dadurch reichern sich Fettstoffe, vor allem "Globotriaosylceramid (GL-3)", in zahlreichen Geweben und Organen an.
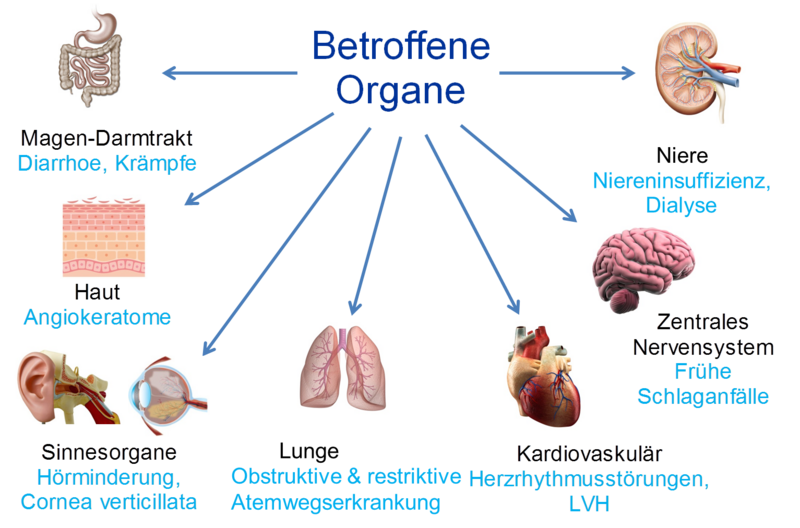
Besonders betroffen sind die Blutgefäße, die Niere und das Herz. Betroffene Organe werden durch diese Ablagerungen geschädigt und ihre Funktion nimmt im Verlauf der Erkrankung ab. An der Haut entwickeln sich rote Flecken, am Auge kommt es zu Ablagerungen in der Hornhaut, im Bereich des Nervensystems können brennende Schmerzen im Bereich der Hände und Füße auftreten, die Schweißdrüsen stellen ihre Funktion ein (vermindertes Schwitzen) und im Bereich des Magen-Darm-Trakts kommt es zu Bauchschmerzen und Durchfall.
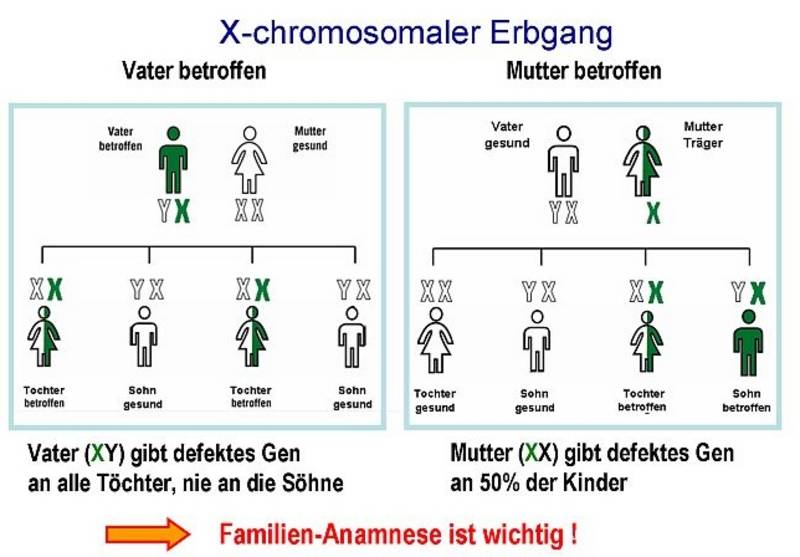
Als Erbkrankheit beruht der Morbus Fabry auf einem Gendefekt auf dem X-Chromosom, dem weiblichen Geschlechtschromosom. Ein Defekt im Gen für die α-Galaktosidase bewirkt, dass der Körper das wichtige Enzym gar nicht oder nicht in ausreichender Menge herstellen kann. Schätzungsweise einer von 40.000 Männern hat einen solchen Gendefekt. Aber auch Frauen sind betroffen. Früher dachte man, dass nur Männer an Morbus Fabry erkranken, da sie nur ein X-Chromosom besitzen. Frauen, die über zwei X-Chromosomen verfügen, galten hingegen lange Zeit nur als Überträgerinnen, die selbst nicht erkranken. Inzwischen weiß man aber, dass auch Frauen an M. Fabry erkranken können, obwohl das Enzym α-GAL bei ihnen häufig noch eine gewisse Restaktivität hat. Bei Männern ist hingegen in der Regel das Enzym im Blut gar nicht nachweisbar. Männer erkranken deshalb meistens schwerer als Frauen und die ersten Symptome treten wesentlich früher auf.
Die Erkrankung "Morbus Fabry" ist seit mehr als 100 Jahren bekannt und doch führt sie bis heute ein Schattendasein. 1898 veröffentlichten zwei Ärzte - "Johann Fabry" aus Deutschland und "William Anderson" aus England - unabhängig voneinander die ersten Berichte über Patienten mit purpurroten punktförmigen Hautflecken, die sich über größere Körperpartien erstreckten.
Damit beschrieben die Ärzte eine der auffälligsten und manchmal auch ersten Veränderungen bei Fabry-Patienten. Inzwischen ist bekannt, dass Morbus Fabry eine Multiorganerkrankung ist, die außer der Haut vor allem die Nieren, das Nervensystem und das Herz betrifft. Deshalb gibt es am Fabry-Zentrum Münster Experten der Fachrichtungen Nephrologie, Neurologie und Kardiologie, die sich speziell mit dem M. Fabry beschäftigen. Da das Krankheitsbild zudem von Patient zu Patient sehr unterschiedlich sein kann, wird eine Fabry-Erkrankung meistens erst mit einer Verzögerung von vielen Jahren erkannt. Es gibt viele Betroffene, die die Ursache ihrer Beschwerden gar nicht kennen. Hier werden die vielfältigen Symptome, die auftreten können, beschrieben und es gibt einen Überblick über die verfügbaren Möglichkeiten zur Behandlung. Seit dem Jahr 2001 steht eine Enzymersatztherapie zur Verfügung.