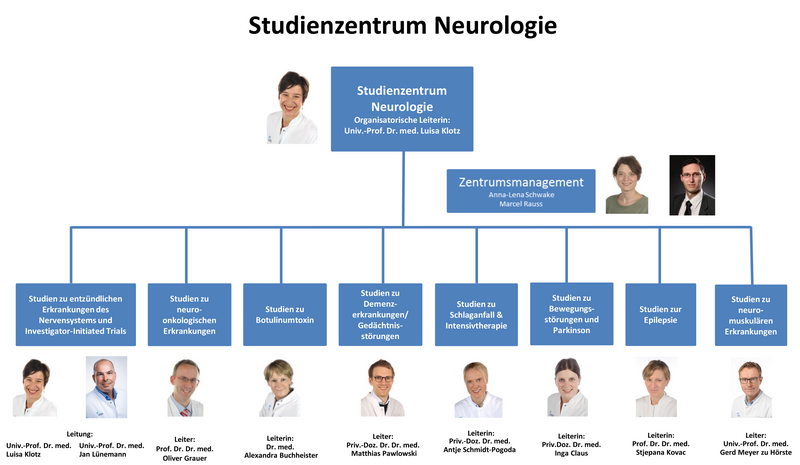
Das Angebot an Studien umfasst die Krankheitsbilder:
Entzündliche Erkrankungen des Nervensystems
Neuroonkologische Erkrankungen
Schlaganfall und andere neurovaskuläre Erkrankungen
Epilepsien


In allen Bereichen der Neurologie werden stetig neue Substanzen und Therapiemethoden entwickelt, mit denen schwere und manchmal noch nicht heilbare Erkrankungen besser behandelt werden können. Die Klinik für Neurologie erforscht diese Therapien und ihre Wirkweise. In unserem Studienzentrum bieten wir eine umfassende Beratung hinsichtlich aktuell verfügbarer Studien als Alternative oder Ergänzung zu bereits zugelassenen Therapieoptionen und betreuen die Patienten intensiv während ihrer Studienteilnahme.
Wollen Wissenschaftler klären, ob eine neue Behandlung sicher und wirksam ist, so tun sie dies meist über klinische Studien. Das gilt für Medikamente ebenso wie für Diäten oder medizinische Geräte (z.B. ein Hirnschrittmacher). Häufig werden dabei verschiedene Therapien miteinander verglichen, um zu sehen, ob eine neue Behandlungsmethode wirksamer ist oder weniger schädliche Nebenwirkungen hat als das Standardvorgehen.
Andere klinische Studien testen Methoden, eine Krankheit frühzeitig zu erkennen, manchmal noch bevor Symptome auftreten. Wieder andere erproben Wege zur Prävention. Eine klinische Studie kann sich auch mit der Frage befassen, wie der Alltag von Menschen verbessert werden kann, die unter einer lebensbedrohlichen Krankheit oder einem chronischen Gesundheitsproblem leiden.
Jede klinische Studie muss vom Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) und der Ethikkommission genehmigt werden. Dazu wird sie einer intensiven Prüfung unterzogen, um stets eine größtmögliche Patientensicherheit zu gewährleisten.
Klinische Studien durchlaufen vier Phasen. In den ersten drei Phasen testen Forscher, ob ein Medikament oder eine andere Intervention sicher und wirksam ist. Ist das der Fall, erfolgt eine Zulassung für den die klinische Anwendung. In Studien der Phase IV werden die Auswirkungen des Medikaments im klinischen Alltag weiterhin überwacht.
Es gibt viele Gründe, warum Menschen an einer klinischen Studie teilnehmen:
Nehmen Sie gerne Kontakt zu uns auf. Wir klären gemeinsam mit Ihnen, ob derzeit eine für Sie passende Studie verfügbar ist und vereinbaren ggf. einen Termin in der Ambulanz.
Sie werden im Vorfeld detailliert über die Studie informiert. Wenn alle Ihre Fragen beantwortet sind und Sie der Teilnahme zustimmen, unterzeichnen Sie eine Einverständniserklärung.
In der anschließenden „Screening-Phase" werden genau untersucht, um sicherzustellen, dass Sie für die Studie geeignet sind. Je nach Studie sind Blutentnahmen, Bildgebungen (MRT oder PET), kognitive oder physische Tests oder auch EKG notwendig. Zu den Einschlusskriterien für eine Studie können aber auch andere Faktoren gehören wie Alter, Krankheitsstadium, Geschlecht, genetisches Profil, Familienanamnese und die Frage, ob Sie einen Studienpartner haben, der Sie bei zukünftigen Besuchen begleiten kann oder nicht.
Daneben gibt es auch Ausschlusskriterien wie spezifische gesundheitliche Probleme oder Medikamente, die die zu prüfende Behandlung beeinträchtigen könnten.
Werden Sie für eine Studienteilnahme zugelassen, findet ein erster Besuch (der „Baseline-Besuch“) statt. Dabei werden Sie meist per Zufallsprinzip einer Behandlungs- oder Kontrollgruppe zugeteilt. Das Medikament wird meist (je nach Studie) als Infusion, Injektion oder als Tablette verabreicht. Es ist wichtig, dass Sie im Verlauf alle Abläufe befolgen und dem Studienteam jederzeit mitteilen, wenn Sie Probleme oder Bedenken bezüglich der Studie haben.
Im Verlauf der Studie müssen Sie zu verschiedenen Terminen ins Studienzentrum kommen, damit Verlaufsuntersuchungen erfolgen können, um die Auswirkung der Studienmedikation und Ihre Gesundheit zu überwachen.
Ein Placebo ist eine Substanz oder Behandlung, die so konzipiert ist, dass sie keinen therapeutischen Wert hat. Bei klinischen Studien kann ein Placebo so gestaltet werden, dass es einem aktiven Medikament oder einer aktiven Therapie ähnelt, so dass es als Kontrolle fungiert. Damit soll verhindert werden, dass der Empfänger oder andere Personen (mit ihrer Zustimmung) wissen, ob eine Behandlung aktiv oder inaktiv ist. Denn die Erwartungen über die Wirksamkeit einer Methode können die Ergebnisse der Studie beeinflussen. Die meisten Studien laufen verblindet ab. Das bedeutet, dass weder der behandelnde Arzt, noch das Studienteam oder der Patient wissen, ob dieser ein Placebo erhält.
Wir sind ein etabliertes Studienzentrum mit langjähriger Erfahrung in einer Vielzahl neurologischer Indikationen, eingebunden in die Strukturen des Universitätsklinikums. Unsere Expertise liegt in der Planung, Organisation und Durchführung klinischer Studien. Unser Leistungsspektrum umfasst Arzneimittel- und Medizinprodukte-Studien der Phasen I bis IV, sowie Anwendungsbeobachtungen.
Alle Studien in der Klinik für Neurologie werden nach den Grundsätzen der Guten Klinischen Praxis (ICH-GCP) durchgeführt. In regelmäßigen Abständen wird unser Personal entsprechend fortgebildet.
Sollten Sie Interesse haben, eine Studie an unserem Zentrum durchzuführen, wenden Sie sich mit Ihrer Anfrage gerne direkt an das Zentrumsmanagement.
Klinische Studien zu komplexen Erkrankungen lassen sich nur umsetzen, wenn Experten über verschiedene Fachgebiete hinweg kooperieren. In dieser interdisziplinären Zusammenarbeit haben wir am Universitätsklinikum Erfahrung. So wird das Team des Studienzentrums von Experten aus der klinikeigenen Apotheke, Nuklearmedizin, Dermatologie, Augenklinik und Radiologie unterstützt. Zudem arbeitet unser Studienzentrum eng mit dem Studienteam der Neurochirurgie zusammen, um interdisziplinäre Vorhaben zu verschiedenen Indikationen, wie beispielsweise die Tiefe Hirnstimulation bei Morbus Parkinson, Tremor oder Dystonien, Intrazerebrale Blutungen oder Glioblastom umzusetzen.
Das Angebot an Studien umfasst die Krankheitsbilder:
Entzündliche Erkrankungen des Nervensystems
Neuroonkologische Erkrankungen
Schlaganfall und andere neurovaskuläre Erkrankungen
Epilepsien

Univ-Prof. Dr. med. Luisa Klotz
Oberärztin

Univ.-Prof. Dr. med. Jan Lünemann
Oberarzt

Marcel Rauss
Finanz- und Drittmittelmanagement
marcel.rauss(at)
ukmuenster(dot)de

Anna-Lena Schwake
Studienanfragen / Studien-Start-up / Koordination
T: +49 2 51 - 83 46081
anna-lena.schwake(at)
ukmuenster.de